We first provide details on how to access the iPSCORE datasets and then below describe the methods used for their generation.
1. How to access iPSCORE datasets
Downloading FASTQ / BAM:
dbGaP download Guide: https://www.ncbi.nlm.nih.gov/sra/docs/sra-dbgap-download/
- To view what raw data is available in phs001325 and phs000924, go to the dbGaP accession page. Click on the “Molecular Datasets'' tab. Then, click on the “Run Selector” link underneath the Table Legend. You may also find sample information in the release notes (https://ftp.ncbi.nlm.nih.gov/dbgap/studies/phs000924/; https://ftp.ncbi.nlm.nih.gov/dbgap/studies/phs001325/).
- To download data, first obtain access to phs001325 for WGS and phs000924 for all other iPSCORE data. You can request access from the dbGaP study page.

- Once access is acquired, navigate to the “Authorized Access Portal” and login. You should see the requested project under the “My Projects” tab.
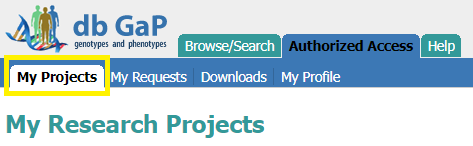
- Get dbGaP repository key from the desired project.
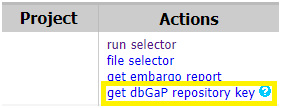
- Extract the SRA Run ID from SraRunTable in the run selector tab. See Step 1 for how to access the SraRunTable. You may also access SraRunTable through your “My Projects” tab after logging into dbGaP (see below).
- Run the following commands to download the sample:
#1 Download SRA file from dbGaP prefetch --ngc ngc_key.ngc SRR1234567 --max-size 420000000000#2.1 To convert SRA to FASTQ: fasterq-dump --ngc ngc_key.ngc --split-files SRR1234567/SRR1234567.sra#2.2 To convert SRA to BAM: sam-dump --ngc ngc_key.ngc SRR1234567.sra | samtools view -b -o path/to/output/location/SRR1234567.bam
*Note: These commands are from SRA Toolkit which is required to download data from dbGaP. Follow the link at the top of this section to the “dbGaP Download Guide” for instructions to install the SRA Toolkit.
Downloading VCF, QTL, SNP, Methylation Array and Summary Statistics Data:
dbGaP download Guide: https://www.ncbi.nlm.nih.gov/sra/docs/sra-dbgap-download
- Before gaining access, you may view what VCF, QTL summary statistics, and Array data are available. Go to the dbGaP accession page. Scroll down to “Authorized Access”. Then, click on the “File Selector” link. You may also find sample information in the release notes (https://ftp.ncbi.nlm.nih.gov/dbgap/studies/phs000924/; https://ftp.ncbi.nlm.nih.gov/dbgap/studies/phs001325/).
- Obtain access to phs001325 for WGS and phs000924 for all other iPSCORE data. You can request access from the dbGaP study page.

- Once access is acquired, get dbGaP accession key
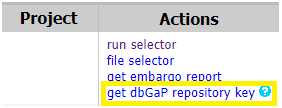
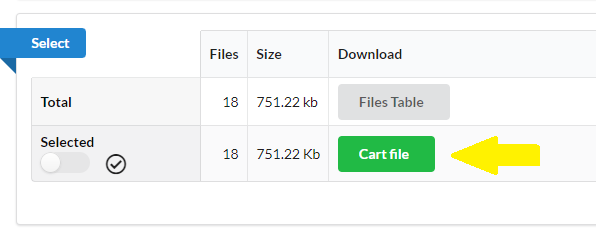
- Select desired files with the file selector and create a cart file
-
Run the following commands to download the sample:
prefetch --ngc ngc_key.ngc {cart_file}.krt --max-size 42000000000
Downloading QTL Summary Statistics from dbGaP Public FTP:
The most recent QTL summary statistics that were uploaded to dbGaP for the Multi-omic QTL and CVPC and PPC eQTL studies are available on dbGaP's public FTP server https://ftp.ncbi.nlm.nih.gov/dbgap/studies/phs001325/phs001325.v6.p1/analyses/FullData/phs001325.iPSCORE_WGS_v6.analysis-PI.MULTI.tar.gz
- This contains all the QTL data and can be downloaded in a browser.
- Once downloaded, it will need to be unzipped and unarchived with the following command:
tar -xzf phs001325.iPSCORE_WGS_v6.analysis-PI.MULTI.tar.gz
2. Methods for iPSCORE datasets
WGS of Blood, Fibroblast, and iPSCs
Genomic DNA was isolated from whole blood (or in 19 cases directly from the fibroblasts) using DNEasy Blood & Tissue Kit (Qiagen), quantified, normalized, and sheared with a Covaris LE220 instrument. The samples were normalized to 1 μg and submitted to Human Longevity (HLI) for whole genome sequencing. DNA libraries were prepared (TruSeq Nano DNA HT kit, Illumina), characterized with regards to size (LabChip DX Touch, Perkin Elmer) and concentration (Quant-iT, Life Technologies), normalized to 2-3.5nM, combined into 6-sample pools, clustered and sequenced to ~50X depth on the Illumina HiSeqX (150 bp paired-end).
Four of the samples were sequenced twice. For the second round of sequencing, genomic DNA was isolated, quantified, normalized, and sheered via the same methods above. 1 μg of material for each sample was submitted to the Institute for Genomic Medicine (IGM) Genomics Center for library preparation and sequencing. DNA libraries were prepared using Kapa Hyper Prep (Roche), qualified for size and purity with a TapeStation 4200 (Agilent) and quantified with a Qubit 2.0 fluorometer (Invitrogen). The libraries were normalized to 10nM and pooled together for sequencing to a depth of ~50X on an Illumina NovaSeq 6000 S4 flow cell (150 bp paired-end).
HumanCoreExome Array
Detailed methods are found in https://doi.org/10.1016/j.stemcr.2017.03.012
Genomic DNA was isolated from iPSC lines (AllPrep DNA/RNA Mini Kit, Qiagen) and from blood or fibroblast germline samples (DNEasy Blood & Tissue Kit), normalized to 200 ng, hybridized in pairs to Illumina HumanCoreExome arrays (Illumina), and stained per Illumina’s standard protocol. The stained Beadchips were then scanned on the Illumina HiScan and processed in GenomeStudio (v 1.9.4).
MEGA Consortium SNP Array
Detailed methods are found in: https://doi.org/10.1016/j.scr.2020.101803
Genotyping was performed through the NHLBI DNA Resequencing and Genotyping (RS&G) Service Center using the Illumina Infinium MEGA Chip. Genotype calls were made using GenomeStudio (version 2011.1) and Genotyping Module (version 1.9.4) and a total of 2,036,060 SNPs for 2,585 samples were released.
DNA Methylation Arrays
Detailed methods are found in: https://doi.org/10.1016/j.stem.2017.03.010
DNA was isolated (DNeasy kit, QIAGEN) from iPSCs at passages 5, 9, and 20 and from fibroblasts at passage 7 and 500 ng was bisulfite converted (EZ DNA Methylation Kit, Zymo Research), of which 250 ng was hybridized to Infinium HumanMethylation450 BeadChip arrays (Illumina). To prevent batch effects, samples from each individual were arrayed across different BeadChips. Methylation levels were processed using the minifi package in R and initially normalized using background subtraction (preprocessIllumina).
Bulk RNA-seq
RNA-seq for iPSCs, CVPCs, and PPCs in iPSC eQTL, NKX2-5, Hi-C, Multi-QTL, X-Chromosome and Time course (iPSC -> CM) studies.
Detailed methods are found in https://doi.org/10.1016/j.xgen.2025.100775.
Total RNA was isolated using AllPrep Mini Kit or Quick-RNA Miniprep Kit. Libraries were prepared using Illumina TruSeq stranded mRNA kit and sequenced with 100 bp paired-end or 150 bp paired-end reads on Illumina HiSeq 2500, HiSeq 4000, or NovaSeq 6000.
RNA-seq for IFNg study
Detailed methods are found in https://doi.org/10.1016/j.jmccpl.2025.100289
We extracted total RNA from lysed pellets frozen in RLT Plus buffer (collected on day 29) using the Quick-RNA™ MiniPrep Kit (Zymo Research), assessed quality to make sure RNA Integrity number (RIN) was 7.5 or greater, prepared indexed libraries using the Illumina TruSeq stranded mRNA kit and sequenced with 150 bp paired-end reads on an Illumina HiSeq 4000 (mean = 91.4M reads).
RNA-seq for CVPC Rybo-Depletion samples (Unpublished)
RNA sequencing libraries were generated using the Illumina TruSeq Stranded Total RNA Library Prep Gold with TruSeq Unique Dual Indexes (Illumina, San Diego, CA). Samples were processed following manufacturer’s instructions, except modifying RNA shear time to five minutes. Resulting libraries were multiplexed and sequenced with 150-bp paired-end reads on an Illumina HiSeq4000. Samples were demuxltiplexed using bcl2fastq v2.20 Conversion Software (Illumina, San Diego, CA).
Bulk ATAC-seq
ATAC-seq for iPSC, CVPC, PPC in Multi-QTL Study
Detailed methods are found in https://doi.org/10.1016/j.xgen.2025.100775.
ATAC-seq samples were processed using a modified version of the Buenrostro et al. protocol. Briefly, frozen nuclear pellets were thawed on ice and tagmented in total volume of 25μl in permeabilization buffer containing digitonin (10mM Tris-HCl pH 7.5, 10mM NaCl, 3mM MgCl2, 0.01% digitonin) and 2.5μl of Tn5 from Nextera DNA Library Preparation Kit (Illumina) for 45-75min at 37°C in a thermomixer (500 RPM shaking). We included a double size selection step during purification using AMPure XP DNA beads (Beckman Coulter). To eliminate confounding effects due to index hopping, libraries within a pool were indexed with unique pairs of i7 and i5 barcodes. Libraries were amplified for 12 cycles using NEBNext® High-Fidelity 2X PCR Master Mix (NEB) in total volume of 25ul in the presence of 800nM of barcoded primers (400nM each) custom synthesized by Integrated DNA Technologies (IDT) and sequenced with either 100 bp paired-end or 150 bp paired-end reads on an Illumina HiSeq4000 or Illumina NovaSeq 6000.
ATAC-seq in NKX2-5 study (PMID 31570892) Hi-C Study (PMID 30837461) and Time course study (iPSC -> CM; unpublished)
Detailed methods are found in https://www.nature.com/articles/s41467-019-08940-5
The ATAC-seq protocol has been adapted from Buenrostro et al. Frozen nuclear pellets of 5 × 104 cells each were thawed on ice, suspended in 50 μL transposition reaction mix (2.5 μL Tn5 transposase in 1× TD buffer, Illumina Cat# FC-121-1030), and incubated for 30 min at 37 °C. Reactions were purified using Qiagen MinElute kit, eluted in 10 μL water and amplified using the KAPA real-time library amplification kit (KAPA Biosystems) with barcoded adaptors. PCR reactions were terminated after 10–13 cycles and purified using AmPure XP beads (Beckman Coulter). Samples were size selected using SPRIselect beads (Beckman Coulter) to a size range of 150–850 kbp and sequenced with 75-bp or 100-bp paired-end reads on an Illumina HiSeq2500.
ATAC-seq in iPSC-RPE study including human fetal RPE
Detailed methods are found in https://www.cell.com/stem-cell-reports/fulltext/S2213-6711.
We performed ATAC-seq using the protocol from Buenrostro et al. (Buenrostro et al., 2013) with small modifications. Frozen nuclear pellets of 5 x 10^4 cells each were thawed on ice and tagmented in permeabilization buffer containing digitonin. Tagmentation was carried in 25ul using 2.5ul of Tn5 from Nextera DNA Library Preparation Kit (Illumina) for 60 min at 37°C in a thermomixer (500 RPM shaking). To eliminate confounding effects due to index hopping, all libraries within a pool were indexed with unique i7 and i5 barcodes. Libraries were amplified for 12 cycles. Libraries were sequenced to approximately 80M 150bp paired end reads on the HiSeq 4000 (Illumina) platform.
ATAC-seq in IFNg study
Detailed methods are found in https://doi.org/10.1016/j.jmccpl.2025.100289
ATAC-seq samples were processed as previously described in detail. Briefly, frozen nuclear pellets of 1×105 CVPC were thawed on ice and tagmented in total volume of 25 μl in permeabilization buffer containing digitonin (10 mM Tris-HCl pH 7.5, 10 mM NaCl, 3 mM MgCl2, 0.01% digitonin) and 2.5 μl of Tn5 from Nextera DNA Library Preparation Kit (Illumina) for 45-75min at 37°C in a thermomixer (500 RPM shaking). To eliminate confounding effects due to index hopping, all libraries within a pool were indexed with unique pairs of i7 and i5 barcodes. Libraries were amplified for 12 cycles using NEBNext® High-Fidelity 2X PCR Master Mix (NEB) in total volume of 25µl in the presence of 800 nM of barcoded primers (400nM each) custom synthesized by Integrated DNA Technologies (IDT) and sequenced with 150-bp paired end reads on a HiSeq4000 (mean = 112.9M reads).
Bulk H3K27ac ChIP-seq
H3K27ac ChIP-seq for iPSC and CVPC in Multi-QTL study
Detailed methods are found in https://doi.org/10.1016/j.xgen.2025.100775.
5-15 x 10^6 formaldehyde crosslinked cells were lysed and sonicated in 110ul of SDS Lysis Buffer (0.5% SDS, 50mM Tris-HCl pH 8.0, 20mM EDTA, 1x cOmplete™ Protease Inhibitor Cocktail (Sigma)) using Covaris E220 Focused-ultrasonicators (Covaris) for 14 cycles, 1 min per cycle, duty cycle 5. For each sample, H3K27ac antibody (Abcam ab4729, lot GR00324078) was coupled for 4 hours to 40ul of 1:1 mix Protein G and Protein A Dynabeads (Thermo Scientific) and used for overnight chromatin immunoprecipitation in IP buffer (1% Triton X-100, 0.1% DOC, 1x TE buffer, 1x cOmplete™ Protease Inhibitor Cocktail). Beads with immunoprecipitated chromatin were washed with 150ul of following buffer: four times with RIPA Low Salt Buffer (0.1% SDS, 1% Triton X-100, 2mM EDTA, 20mM Tris-HCl pH 8.0, 300mM NaCl, 0.1% DOC), two times in RIPA High Salt Buffer (0.1% SDS, 1% Triton X-100, 1mM EDTA, 20mM Tris-HCl pH 8.0, 500mM NaCl, 0.1% DOC), twice in LiCl Buffer (250mM LiCl, 0.5% NP-40, 0.5% DOC, 1mM EDTA, 10mM Tris-HCl pH 8.0) and twice in 1X TE buffer (10mM Tris-HCl pH 8.0, 1mM EDTA). Next samples were eluted in 150 ul of Direct Elution Buffer (0.1% SDS, 10mM Tris-HCl pH 8.0, 5mM EDTA) and reverse crosslinked by incubation for 15 min at 65˚C with rotation and subsequent incubation with 5 ul RNAse (Sigma) for 1h at 37˚C and Proteinase K Solution (20 mg/mL, Thermo Fisher Scientific) for 1h at 55˚C. After reverse crosslinking samples were purified with 2X Agencourt AMPure XP DNA beads (Beckman Coulter), eluted in 30 ul of H2O and Qubit (Thermo Scientific) quantified. Libraries were generated using KAPA Hyper Prep Kit (KAPA Biosystems) and KAPA Real Time Library Amplification Kit (KAPA Biosystems) following manufacturers manual. Libraries were barcoded using TruSeq RNA Indexes (Illumina), size selected for 300 bp to 500 bp, and sequenced with either 100 bp paired-end or 150 bp paired-end reads on an Illumina HiSeq 4000.
Control Input H3K27ac ChIP-seq for iPSC and CVPC in Multi-QTL Study
Detailed methods are found in https://doi.org/10.1016/j.xgen.2025.100775.
For control input chromatin, 5-15 x 106 formaldehyde crosslinked cells were lysed and sonicated in 110µl of SDS Lysis Buffer (0.5% SDS, 50mM Tris-HCl pH 8.0, 20mM EDTA, 1x cOmplete™ Protease Inhibitor Cocktail (Sigma)) using Covaris E220 Focused-ultrasonicators (Covaris) for 14 cycles, 1 min per cycle, duty cycle 5. Input chromatin was then reverse crosslinked by incubation for 15 min at 65˚C with rotation and subsequent incubation with 5 µl RNAse (Sigma) for 1h at 37˚C and Proteinase K Solution (20 mg/mL, Thermo Fisher Scientific) for 1h at 55˚C. After reverse crosslinking, samples were purified with 2X Agencourt AMPure XP DNA beads (Beckman Coulter), eluted in 30 µl of H2O and Qubit (Thermo Scientific) quantified. Libraries were generated using KAPA Hyper Prep Kit (KAPA Biosystems) and KAPA Real Time Library Amplification Kit (KAPA Biosystems) following manufacturers manual. Libraries were barcoded using TruSeq RNA Indexes (Illumina), size selected for 300 bp to 500 bp, and sequenced with either 100 bp paired-end or 150 bp paired-end reads on an Illumina HiSeq 4000.
H3K27ac ChIP-seq for CM in NKX2-5 and Hi-C Studies (PMIDs 31570892 30837461)
Detailed methods are found in https://www.nature.com/articles/s41467-019-08940-5#Sec15.
For H3K27ac, 2 × 106 fixed cells were lysed in 60 ul of MAGnify™ Chromatin Immunoprecipitation System Lysis Buffer (Thermo Scientific) and sonicated using Bioruptor 200 (Diagenode) for 35–45 min of 30 s on/30 s off cycles. H3K27ac antibodies (Abcam ab4729, lots GR183922-2 (1.75 ug) or GR184333-2 (1 ug)) were coupled for 2 h to ProteinG Dynabeads (Thermo Scientific), and used for overnight chromatin immunoprecipitation in IP buffer (1% Triton-X, 0.1% DOC, 1× TE, 1× Roche Complete Proteinase Inhibitor tablets (RCPI)). Beads were washed five times with washing buffer (50 mM Hepes pH 8, 1% NP-40, 0.7% DOC, 0.5 M LiCl, 1 mM EDTA, and 1× RCPI) and once with TE buffer. DNA was eluted and reverse crosslinked overnight in elution buffer (10 mM Tris-HCl pH 8, 1 mM EDTA, 1% SDS) at 65 °C. DNA was purified using Qiagen MinElute PCR Purification kit, quantified by Qubit (Thermo Scientific) and submitted to library preparation and barcoding using KAPA Hyper Library preparation kit (KAPA Biosystems). Libraries were sequenced on an Illumina HiSeq2500 or a HiSeq4000 to an average of 35 M 100 bp paired-end reads per sample.
H3K27ac ChIP-seq for iPSC-RPE and human fetal RPE
Detailed methods are found in https://www.cell.com/stem-cell-reports/fulltext/S2213-6711.
For H3K27ac, 5-15 x 10^6 formaldehyde crosslinked cells were lysed and sonicated in 110 ul of SDS Lysis Buffer (0.5% SDS, 50 mM Tris-HCl pH 8.0, 20 mM EDTA, 1x cOmplete™ Protease Inhibitor Cocktail (Sigma)) using Covaris E220 Focused-ultrasonicators (Covaris) for 14 cycles, 1 min per cycle, duty cycle 5. For each sample, H3K27ac antibody (Abcam ab4729, lot GR00324078) was coupled for 4 hours to 20 ul of Protein G Dynabeads (Thermo Scientific) and used for overnight chromatin immunoprecipitation in IP buffer (1% Triton X-100, 0.1% DOC, 1x TE buffer, 1x cOmplete™ Protease Inhibitor Cocktail). Beads with immunoprecipitated chromatin were washed six times with 150 ul of ChIP wash buffer (50 mM HEPES pH 8.0, 1% NP-40, 0.7% DOC, 400 mM LiCl, 1 mM EDTA, 1x cOmplete™ Protease Inhibitor Cocktail) and twice with 1X TE buffer (10 mM Tris-HCl pH 8.0, 1 mM EDTA). Next, samples were eluted in 150 ul of ChIP Elution Buffer (1% SDS, 10 mM Tris-HCl pH 8.0, 1 mM EDTA) and reverse crosslinked by incubation for 15 min at 65˚C with rotation and subsequent incubation with 5 ul RNAse (Sigma) for 1h at 37˚C and Proteinase K Solution (20 mg/mL, Thermo Fisher Scientific) for 1h at 55˚C. After reverse crosslinking, samples were purified with 2X Agencourt AMPure XP DNA beads (Beckman Coulter), eluted in 30 ul of H2O, and quantified using Qubit (Thermo Scientific). Libraries were generated using KAPA Hyper Prep Kit (KAPA Biosystems) and KAPA Real-Time Library Amplification Kit (KAPA Biosystems) following the manufacturer's manual. Libraries were barcoded using TruSeq RNA Indexes (Illumina) and sequenced on an Illumina HiSeq 4000 to an average of 40 M 150 bp paired-end reads per sample.
Control Input H3K27ac ChIP-seq for iPSC-RPE and human fetal RPE
For H3K27ac, 5-15 x 10^6 formaldehyde crosslinked iPSC-RPE cells were lysed and sonicated in 110 ul of SDS Lysis Buffer (0.5% SDS, 50 mM Tris-HCl pH 8.0, 20 mM EDTA, 1x cOmplete™ Protease Inhibitor Cocktail (Sigma)) using Covaris E220 Focused-ultrasonicators (Covaris) for 14 cycles, 1 min per cycle, duty cycle 5. Next, samples were reverse crosslinked by incubation for 15 min at 65˚C with rotation and subsequent incubation with 5 ul RNAse (Sigma) for 1h at 37˚C and Proteinase K Solution (20 mg/mL, Thermo Fisher Scientific) for 1h at 55˚C. After reverse crosslinking, samples were purified with 2X Agencourt AMPure XP DNA beads (Beckman Coulter), eluted in 30 ul of H2O, and quantified using Qubit (Thermo Scientific). Libraries were generated using KAPA Hyper Prep Kit (KAPA Biosystems) and KAPA Real-Time Library Amplification Kit (KAPA Biosystems) following the manufacturer's manual. Libraries were barcoded using TruSeq RNA Indexes (Illumina) and sequenced on an Illumina HiSeq 4000 to an average of 40 M 150 bp paired-end reads per sample.
H3K27ac ChIP-seq Timecourse (iPSC -> CVPC) (Unpublished)
2 × 106 fixed cells were lysed in 60 ul of MAGnify™ Chromatin Immunoprecipitation System Lysis Buffer (Thermo Scientific) and sonicated using Bioruptor 200 (Diagenode) for 35–45 min of 30 s on/ 30 s off cycles. H3K27ac antibodies (Abcam ab4729, lots GR183922-2 (1.75 ug) or GR184333-2 (1 ug)) were coupled for 2 h to ProteinG Dynabeads (Thermo Scientific), and used for overnight chromatin immunoprecipitation in IP buffer (1% Triton-X, 0.1% DOC, 1× TE, 1× Roche Complete Proteinase Inhibitor tablets (RCPI)). Beads were washed five times with washing buffer (50 mM Hepes pH 8, 1% NP-40, 0.7% DOC, 0.5 M LiCl, 1 mM EDTA, and 1× RCPI) and once with TE buffer. DNA was eluted and reverse crosslinked overnight in elution buffer (10 mM Tris-HCl pH 8, 1 mM EDTA, 1% SDS) at 65 °C. DNA was purified using Qiagen MinElute PCR Purification kit, quantified by Qubit (Thermo Scientific) and submitted to library preparation and barcoding using KAPA Hyper Library preparation kit (KAPA Biosystems). Libraries were sequenced with 100-bp paired-end reads on an Illumina HiSeq2500.
Hi-C
Hi-C for iPSC and CM
Detailed methods are found in https://doi.org/10.1038/s41467-019-08940-5.
Hi-C libraries were prepared using in situ Hi-C. Cells were crosslinked at a final concentration of 1% formaldehyde and quenched using 200 mM glycine. Crosslinked cells were then lysed and nuclei were digested with 100 U MboI overnight at 37 °C. Next, fragmented ends were biotinylated for 90 min at 37 °C, and the sample was diluted and proximity ligated for 4 h at room temperature. Crosslinks were reversed by the addition of SDS, ProteinaseK, and NaCl, and allowed to incubate overnight at 68 °C. Samples were then purified by ethanol precipitation, resuspended in 100 µL 1× Elution Buffer, fragmented using a Covaris S2 instrument, and size selected using AmpureXP beads. Subsequently, biotinylated ligation junctions were pulled down using T1 Streptavidin beads. Hi-C libraries were prepared using streptavidin beads by performing end-repair, dA-tailing, and adapter ligation, following which PCR amplification and purification was performed. The resulting libraries were sequenced on an Illumina HiSeq 4000 machine to obtain 150 bp paired-end reads.
Single-cell RNA-seq
CVPC scRNA-seq
Detailed methods are found in https://doi.org/10.1016/j.stemcr.2019.09.011.
Single cells were captured using the 10x Chromium controller (10x Genomics) according to the manufacturer’s specifications and manual (Manual CG00052, Rev C). Cells for each sample were loaded on the individual lane of a Chromium Single Cell A Chip. Libraries were generated using Chromium Single Cell 3’ Library Gel Bead Kit v2 (10xGenomics) following manufactures manual. Libraries were sequenced using a custom program (26-8-98 Pair End) on HiSeq 4000. Each library was sequenced on an individual lane. In total we captured 36,839 cells. We retrieved FASTQ files and used CellRanger V2.1 (https://support.10xgenomics.com/) with default parameters using Gencode V19 gene annotation to generate single-cell gene counts for each individual sample.
PPC scRNA-seq
Detailed methods are found in https://doi.org/10.1038/s41467-023-42560-4.
All single cells were captured using the 10X Chromium controller (10X Genomics) according to the manufacturer’s specifications and manual (Manual CG000183, Rev A). Cells from each scRNA-seq sample (one iPSC, seven fresh iPSC-PPCs, RNA_Pool_1, and RNA_Pool_2) were loaded each onto an individual lane of a Chromium Single Cell Chip B. Libraries were generated using Chromium Single Cell 3’ Library Gel Bead Kit v3 (10X Genomics) following manufacturer’s manual with small modifications. Specifically, the purified cDNA was eluted in 24 μl of Buffer EB, half of which was used for the subsequent step of the library construction. cDNA was amplified for 10 cycles and libraries were amplified for 8 cycles. All libraries were sequenced on a HiSeq 4000 using custom programs (fresh: 28-8-175 Pair End and cryopreserved: 28-8-98 Pair End). Specifically, eight libraries generated from fresh samples (one iPSC and seven iPSC-PPC samples) were pooled together and loaded evenly onto eight lanes and sequenced to an average depth of 163 million reads. The two libraries from seven cryopreserved lines (RNA_Pool_1 and RNA_Pool_2) were each sequenced on an individual lane to an average depth of 265 million reads.
Single-nuclei ATAC-seq
PPC snATAC-seq
Detailed methods are found in https://doi.org/10.1101/2021.10.20.465206.
A total of 7 iPSC-PPC samples were used for snATAC-seq generation. Cells from seven cryopreserved iPSC-PPCs samples were captured for snATAC-seq immediately after thawing. All seven samples have matched scRNA-seq. Prior to capture, cells from four cryopreserved iPSC-PPC samples were pooled (ATAC_Pool_1) and cells from the other 3 iPSC-PPC samples were pooled without a freezing step (ATAC_Pool_2). Nuclei from the two pools were isolated according to the manufacturer’s recommendations (Manual CG000169, Rev B), transposed, and captured as independent samples according to the manufacturer’s recommendations (Manual CG000168, Rev B). All single nuclei were captured using the 10x Chromium controller (10x Genomics) according to the manufacturer’s specifications and manual (Manual CG000168, Rev B). Cells for each sample were loaded on the individual lane of a Chromium Chip E. Libraries were generated using Chromium Single Cell ATAC Library Gel & Bead Kit (10x Genomics) following manufacturer’s manual (Manual CG000168, Rev B). Sample Index PCR material was amplified for 11 cycles. Libraries were sequenced using a custom program (50-8-16-50 Pair End) on HiSeq 4000. Specifically, two libraries from seven cryopreserved iPSC-PPC samples (ATAC_Pool_1 and ATAC_Pool_2) were each sequenced on an individual lane.
Bulk NKX2-5 ChIP-seq
NKX2-5 ChIP-seq for CM in NKX2-5 Study
Detailed methods are found in the supplemental notes: doi: 10.1038/s41588-019-0499-3
For NKX2-5, 1-2 x 107 cells were lysed in 300 µl RIPA buffer (1xPBS, 1% NP-40, 0.5% DOC, 0.1% SDS, RCPI) and sonicated for 70-80 min using Bioruptor 200 with instrument and setting as above. Five µg of NKX2-5 antibody (Santa Cruz Biotechnology, sc-8697x, lot C0113) were incubated with Dynabeads for 2 hours and washed with BSA 0.5% in PBS. Chromatin was diluted to 1 ml of RIPA buffer and added to the beads for overnight IP. Five washes were performed with washing buffer (50 mM Hepes pH 8, 1% NP-40, 0.7% DOC, 0.5M LiCl, 1mM EDTA and 1x RCPI), followed by one wash with TE. DNA was eluted 2 and reverse crosslinked in elution buffer (10 mM Tris-HCl pH 8, 1 mM EDTA, 1% SDS) at 65˚C, followed by RNAse A (Sigma) and Proteinase K (Thermo Scientific). DNA was purified using Qiagen MinElute PCR Purification kit (or using 2X Agencourt AMPure XP DNA beads, Beckman Coulter), quantified by Qubit (Thermo Scientific) and submitted to library preparation and barcoding using KAPA Hyper Library preparation kit (KAPA Biosystems). Libraries were sequenced on an Illumina HiSeq2500 or a HiSeq4000 to an average of 35 M 100 bp paired-end reads per sample.
CTCF ChIP-seq
CTCF for iPSCs
Detailed methods are found in https://doi.org/10.1016/j.stem.2017.03.009.
Cells were cross linked with 1% formaldehyde for 10min at room temperature. For each sample 3x10ˆ6 cells were lysed and sonicated using Covaris M220 for 10min. Sonicated chromatin were immunoprecipitated with anti-CTCF antibody (Santa Cruz Biotechnology, sc-15914 X). Libraries were prepared using Illumina TruSeq adapters, size selected for 300bp-500bp, and sequenced on Illumina HiSeq4000 for paired-end sequencing to obtain 100bp reads. Samples were sequenced to an average of ∼44 million read pairs.